|
 |
 |
 |
 |
 |
 |
- Strategies for novel nanoporous materials based on supramolecular coordination chemistry
- New materials for selective catalysts
- New materials for selective membrane separations
- Nanoporous materials for energy storage
- Development of multiscale modeling techniques
- Use of modeling to guide development of new materials
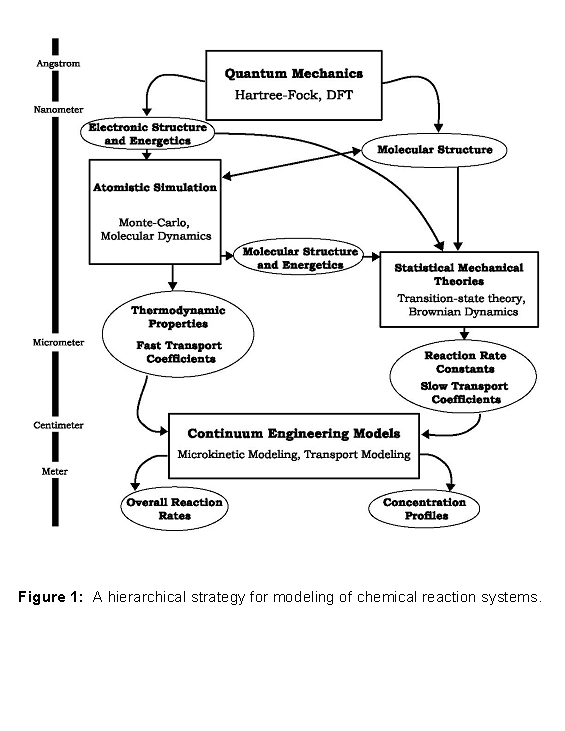
Multiscale Modeling. This project is developing multi-scale modeling approaches and applying them to novel nanoporous materials for selective oxidation catalysis, membrane separations, and energy storage. The modeling approach spans hierarchically from the atomic level, through mesoscale domains, and up to macroscopic scales. See Figure 1 below. Much progress has already been made in developing modeling techniques appropriate for various time and length scales, e.g. quantum chemistry approaches at the atomic level, molecular dynamics simulations for length scales of hundreds of Angstroms, and engineering models at macroscopic scales. The grand challenge - essential for solving complex, integrated problems - is to effectively link these different techniques into a comprehensive hierarchical approach. We are working to do this through several novel developments. First, state-of-the-art embedded cluster quantum chemical methods are being interfaced with iterative quantum/classical calculations. We have also developed a way of incorporating low-level electronic-structure information into statistical mechanical simulations through a novel use of the Fukui function. To perform modeling at reactor scales, we are developing means to link finite-difference models, kinetic Monte Carlo, and microkinetic reaction modeling. The reactor-scale modeling consolidates results from the other calculations in the hierarchy by using calculated reaction information and diffusion coefficients as inputs.
Catalysis for Green Chemistry and Engineering. A diverse set of skills are required to make real progress in the difficult task of creating high-tech catalysts. What are the properties of an "ideal" catalyst? In our opinion they are the following: all sites should be identical; the sites should be tailored to achieve high activity and selectivity; the catalyst should not deactivate; and there should be good mass transport of reactants and products to and from the active sites. Achieving this requires skills in diverse areas including catalyst synthesis and characterization; reaction kinetic studies; mass transport studies; and structure/property relationships (i.e. modeling) to guide the tailoring. Working together, our team members have these skills, and we are developing novel "molecular squares" as hosts for catalytic sites that would have the required properties.
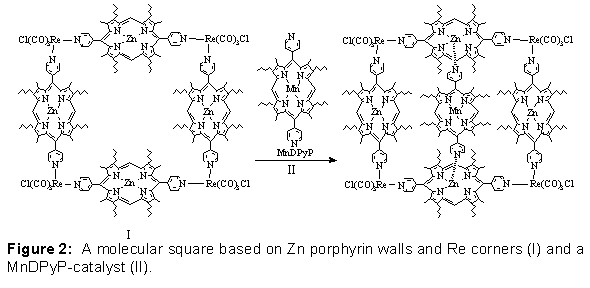
One target is to substantively amplify the performance of new nanoporous-materials-based "artificial enzymes" devised by the Nguyen and Hupp groups using "molecular squares" as building blocks. Briefly, the molecular square catalysts feature highly potent, but inherently unstable biomimetic catalysts (II in Figure 2, below), which are protectively encapsulated in designed high-symmetry cavities of nanometer dimension to stabilize them (I in Figure 2, below). The cavities, in turn, may be self-organized into channels, which are further organized into periodic arrays such that the resulting macroscopic materials display accelerated molecular transport behavior. In contrast to the isolated catalysts, the encapsulated assemblies are much less prone to deactivation, and they have the potential for enzyme-like reactivity, i.e. an ability to transform - in a completely catalytic fashion - targeted reactant molecules to desired products in a shape-specific, size-specific, and enantio-specific manner. With our closely-coupled experimental/modeling approach, we seek to truly design and synthesize nanoscale cavity environments that include hundreds of atoms to create the effects that we desire, rather than relying on the catalytic power of just a small active site. Then, through self-assembly, these cavities can be organized on length scales of tens to thousands of nanometers.
A Few Results. Recently we showed that thin films of molecular squares can be used as membranes for size-selective separations [M.H. Keefe, J.L. O'Donnell, R.C Bailey, S. T. Nguyen, J. T. Hupp, Adv. Mater. 15, 1936-1939 (2003).; J.L. O'Donnell, M.H. Keefe, and J.T. Hupp, Mat. Res. Soc. Symp. Proc. 734, B1.1.1-B1.1.12 (2002).; K.F. Czaplewski, J.T. Hupp, R.Q. Snurr, Adv. Mater. 13, 1895-1897 (2001).] Combination of this membrane separation capability with catalytic reaction is a current goal. The combination of modeling and experiment has already proven its usefulness. For example, in their crystalline form, squares with pyrazine edges exhibit a regular channel system running between the squares but it is not clear if molecules will fit through the pores of this, the smallest, molecular square. Simple molecular models helped differentiate the role of diffusion of molecules through the cavities of the molecular squares versus diffusion between the squares.
The ability of molecular squares to significantly improve the stability and selectivity over bare porphyrin catalysts by encapsulating them was recently reported [M.L. Merlau, M. del Pilar Mejia, S.T. Nguyen, J.T. Hupp, Angew. Chem. Int. Ed. 40, 4239-4242 (2001)]. This demonstration of the "artificial enzyme" nature of the systems was carried out for epoxidation reactions of olefins. The lifetime of the catalyst was improved from 59 turnovers for the bare catalyst to 490 for the protectively encapsulated system. It can be hypothesized that thin films of these materials will demonstrate lifetimes many orders of magnitude longer than this, as the catalytic sites will be unable to desorb into solution as readily, preventing their deactivation by oxo-dimer formation. The encapsulated catalysts also showed enhanced selectivity, for example converting less bulky reactants 7 times faster than bulkier reactants.
Please see the publications page for more information on our current activities.