The overarching motif of research in our group is the synthesis and characterization of metal oxides and oxide fluorides for materials and catalysis applications. Metal oxides are abundant in nature and exhibit high chemical and thermal stability so they have found a ubiquitous commercial applications ranging from advanced electronics to clean energy production to catalysis. We aim to understand the fundamental chemical processes that drive the formation of a particular crystal structure, which exhibits the electrical, optical and/or magnetic properties of interest. Furthermore, we develop synthesis methods to control the particle size and shape for catalytic applications. Our synthesis techniques target large single crystals, polycrystalline powders and single-crystal nanoparticles to determine the fundamental crystal structure and test the material for practical applications. We use molten fluxes, floating zone furnaces, solid state reactions, hydrothermal synthesis and microwave-assisted synthesis. The research spans five collaborative centers, which allows individual projects to grow and develop in neighboring fields. We target materials that have applications as frequency-doubling crystals, transparent conducting oxides, photovoltaic efficiency layers, hydrogen fuel cell anodes, multivalent battery cathodes, catalysts and catalytic supports.
The arsenal of synthetic methods for solid-state chemistry includes the direct combination high-temperature approaches, synthesis from fluxes and melts, hydrothermal synthesis, and synthesis from solutions.
Solid State Reactions - Traditional solid state chemistry reacts constituent powders to form complex crystal structures. Because of the very low diffusion coefficients in solids (on the order of 10-12 cm2s-1) high temperatures over 1000 °C are necessary. Solid state reactions have a long history in inorganic chemistry and ceramics and are popular in industry owing to their simplicity and reproducibility.
Hydrothermal Synthesis - Hydrothermal syntheses involve chemical reactions in water above ambient temperature and pressure in a sealed or closed system and are a special type of chemical transport reaction that relies on liquid phase transport of reactants to nucleate formation of the desired product. Under autogenous conditions, water functions both as a pressure transmitting medium and as a solvent. In a sealed vessel, the vapor pressure of water increases as the temperature is raised above its normal boiling temperature, but below its critical temperature. The selected reaction temperature and the degree of fill, or the percent of the reaction vessel free volume that is filled with water at room temperature, determine the prevailing experimental pressure. When using water as a solvent the dielectric constant and viscosity are also important. These decrease with rising temperature and increase with rising pressure, the temperature effect predominating. Owing to the changes in the dielectric constant and viscosity of water, the increased temperature within a hydrothermal medium has a significant effect on the speciation, solubility and transport of solids.
If elevated temperatures alone are insufficient to dissolve the solid reagents, then a mineralizer can be added, whose ions form complexes with the solids and render them more soluble. These solubilizing reagents include acids, bases and alkali salts. We use one of the aforementioned types of mineralizers in almost every hydrothermal reaction we study. Our transparent conducting oxides (TCOs) are synthesized under basic conditions and our reactions yielding oxide fluorides use either hydrofluoric acid or an alkali fluoride as the mineralizer.
With standard PTFE Teflon-lined stainless steel pressure vessels, one experiment per vessel can be performed, and exploratory synthesis would require numerous vessels and consume a significant amount of time. To overcome this obstacle, we use semi-combinatorial exploration to rapidly gain understanding of a chemical reaction under hydrothermal conditions. Multiple FEP Teflon pouches are employed as individual reaction vessels placed within a single pressure vessel of significantly larger size. The sealed pouches are semi-permeable to water and air under reaction conditions, but not to the solid materials. Thus, a specific reaction can easily be studied by changing one variable across all pouches without being concerned about possible differences in reaction conditions.
Molten Flux Solvents - Synthesis from fluxes can be viewed as slow cooling of a melt that can have a composition very different from the resulting crystalline phase. This technique is popular for crystal growth of oxides. Selection of a suitable metal as a flux medium is based on a number of criteria such as nonzero solubility of the reagents, low melting point, and inertness. The reagent solubility provides high reactivity while the low melting point allows for kinetic control over the reaction and eventual isolation of new phases. Removal of the flux is done in a number of ways such as chemical etching with various solutions or mechanical decanting/centrifuging at elevated temperature while it is still molten.
Microwave Synthesis - The application of temperature to a reaction typically utilizes an external heating source, such as a ceramic coil, to heat the air surrounding the reaction. Microwaves, however, heat the water or reaction medium directly, which decreases the reaction times and increases temperature control of a reaction. Furthermore, high monodispersity of particle size and shape has been observed as well as the formation of alternative products compared to hydrothermal syntheses.
Noncentrosymmetric solids, or materials without any centers of symmetry, exhibit important dielectric and elastic properties, including ferro-, pyro- or piezoelectricity, ferroelasticity and optical activity. An example of an application of noncentrosymmetric materials is frequency-doubling crystals for lasers. Polar distortions in metal centered octahedra are believed to be the origin of the nonlinear optical response in metal oxides. Octahedrally coordinated d0 transition metal cations in Groups 4 and 5 such as Ti4+ and Nb5+ are unstable in mixed metal oxides, and tend to undergo a distortion in which the transition metal cation moves away from the center of its octahedron. While the noncentrosymmetric crystal classes required for solids to exhibit these properties have been mathematically derived and are well known, the structural design principles that facilitate the supramolecular assembly of materials with specific chemical properties are less well defined.
In our work, first we are carrying out a series of syntheses aimed specifically at elucidating the fundamental behavior of [MOxF6-x]n- anions in purely inorganic, solid-state environments. Second, we are also studying the effects of crystallizing the oxide fluoride anions with multiple, different alkali cations. Finally, we are exploring the use of HF(aq) as a fluoride source and mineralizer.
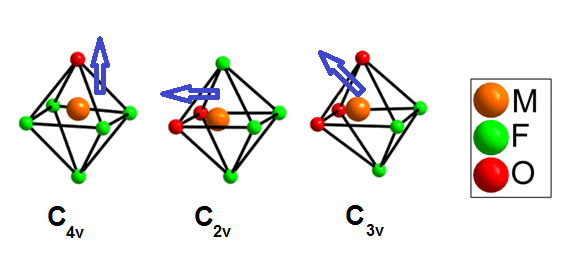
The out-of-center distortions present in [MOxF6-x]n- anions result from both the inherent differences in the nature of M-O and M-F bonding and, to a lesser degree, from electrostatic interactions between the anions and the extended structure of the material. Here, the 2p valence orbitals of the oxide ligands mix with the transition metal d orbitals more strongly than the fluoride orbitals because the energy of the fluoride valence orbitals is extremely low. The result is an out-of-center movement of the central cation towards the oxide ligand(s) that is inherent to each [MOxF6-x]n- anion. This "primary" distortion can be directed toward a corner, edge, or face of the octahedron, depending on the number of oxide ligands (one, two, or three, respectively) that are coordinated to the transition metal cation. In our work, we have synthesized via high-throughput hydrothermal conditions, inorganic solid state compounds with the [NbOF5]2- anion. Instead of replacing cations of a single alkali metal with [NbOF5]2- anions, however, mixed-metal cation systems have been employed to create multiple coordination environments around the oxide and fluoride ligands to aid in the crystallographic ordering of the anion. Two such compounds include KNaNbOF5 and CsNaNbOF5. Isolation of the [NbOF5]2- anion in these two compounds has allowed us to characterize primary and secondary distortions in a purely inorganic, solid-state environment. Recently, we have synthesized mixed alkali cation compounds that contain the [MoO2F4]2- and [WO2F4]2- anions.
Oxide - fluorides developed in our group have also found an application as Li battery cathodes. Implantable medical devices are used to address a variety of medical needs including nerve stimulation, drug delivery, bone growth, and cardiac management. One specific device, the internal cardioverter defibrillator (ICD), monitors the patient's heart in order to deliver therapy when the heart experiences tachycardia. For this, high power from a primary lithium battery needs to quickly charge two capacitors with as high as 30 - 40 J at 700 - 800 V which will be discharged into the heart. The present industry standard in ICDs is a primary lithium battery composed with a cathode of silver vanadium oxide Ag2V4O11 (SVO) owing to its ability to provide a high current at high potential (above 3 V) associated to the reduction of silver. The synthesis of the new material Ag4V2O6F2 (SVOF) and the subsequent electrochemical characterization shows that the higher density of silver within the phase and the inclusion of the more electronegative fluoride provide about a 50% higher gravimetric capacity above 3 V and delivered at 300 mV greater than SVO. Owing to the higher potential (faster capacitor charge time) and higher silver density, there is significant commercial interest in using this material for lithium batteries in ICDs. The technical objective of this research is to improve upon the synthesis of SVOF toward smaller particles and scaled up quantities and to work with the interested industrial parties.
This material is based upon work supported by the National Science Foundation under Grant Nos. DMR-1608218, DMR-1307698, DMR-1005827, DMR-0604454, DMR-0312136.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Transparent conducting oxides (TCOs) are crucial materials to many commercial applications, including flat-panel displays, liquid crystal displays, photovoltaic devices, light emitting diodes and energy efficient windows. TCOs exhibit both optical transparency in the visible spectrum and electrical conductivity - two properties that are typically mutually exclusive. Few compounds exist that satisfy the electronic requirements, in particular, d10 post-transition metal oxides, In2O3, SnO2 and ZnO. Tin-doped indium oxide (ITO) exhibits the highest conductivities and transparencies, but the high cost and scarcity of indium metal has driven research towards developing alternative materials. Our group synthesized In2-2xZnxSnxO3 (ZITO), which uses less indium than ITO and has collaborated with other groups to test the performance in devices and begin to understand the conduction mechanism.
The commercial application of TCOs in currently limited to n-type conductors because the few known p-type conductors are too poor for devices. A major challenge in TCO research is developing p-type conductors that exhibit both high conductivities and optical transparencies. The conductivities of known p-type TCOs are at least 100 times lower than those of the n-type TCOs.
One class of p-type TCOs is the delafossite structure, which follows the general formula ABO2, where A is monovalent and B is trivalent. We have a devised a low temperature (< 210 °C) and low pressure (< 20 atm) hydrothermal technique to synthesize all the known copper CuBO2 (B = Al, Sc, Cr, Mn, Fe, Co, Ga and Rh) and silver AgBO2 (B = Al, Sc, Fe, Ga, Co, Ni, Rh, In and Tl) delafossite oxides in moderate to high yields. In contrast to two-step ion-exchange syntheses, or very high pressure syntheses, all the known silver delafossites have been synthesized by a convenient, practical and direct reaction for the first time, including several key compositions AgAlO2, AgGaO2, and AgScO2, which have never been synthesized in any practical (good yield) reaction before. Similar to the synthesis of the simple delafossite-type oxides, initial research shows that the solubility of the A- and B-site cations play a critical role in product formation of solid solutions. At the current time, we have synthesized the following solid solutions: Ag(Ga,Al)O2, Ag(Sc,In)O2, and Cu(Ga,Al)O2. A difference in B-site cation solubility ≥ 10-3 M renders the synthesis of that particular solid-solution inaccessible by our hydrothermal technique. These results are preliminary and the hope is that by altering the mineralizer concentration (pH), we will be able to expand upon this list.
We are currently collaborating with theorists and materials scientists to approach the search for a p-type TCO from an innovative perspective. Traditional materials research starts with the discovery of a new compound (or realizing a new application for an old compound). After the physical properties have been measured, the origin of the properties and electronic structure is developed by theorists. The novel approach for discovering new compounds starts by defining the theoretical target electronic structure and properties. A theoretical structure and composition are then optimized to predict high-performance compounds. We then apply our techniques to synthesize the compound in high yield. Current research is starting with alternative model structures such as dipolar Ag3VO4-type or mixed anion, i.e., oxysulfide or oxyfluoride.
Our research has recently moved into synthesizing solar energy components other than the TCO layer (anode). A simple photovoltaic device requires only an anode, absorbing layer and cathode. Multiple electronic processes, however, occur in these materials, so additional layers are added to direct the flow of electrons and improve device efficiency. We are developing a solution-based method to grow thin films of p-type oxides, which act as an electron blocking layers. We are starting with NiO, which has been demonstrated to improve device efficiency. The electron blocking layer is inserted between the TCO and the absorbing layer to allow holes and prevent electrons from moving to the anode.
Collaborative Centers
Materials Research Science and Engineering Center (http://www.mrsec.northwestern.edu/)
Center for Inverse Design (http://centerforinversedesign.org/)
Argonne-Northwestern Solar Energy Research Center (http://www.ansercenter.org/)
The focus of this research is to determine the surface structure of multi-component metal oxides and understand the reactions occurring on their surfaces. To reach this objective, single crystals with well-defined crystallographic orientations are needed. We study data from XPS, UV Raman, and TEM to determine connectivity and coordination environments. This information can be used to elucidate a reductive addition and oxidative elimination pathway. Our group targets catalysts for functionalization of simple hydrocarbons as well as conversion of biomass into more reactive molecules.
Perovskite oxide surfaces have been used as heterogeneous catalysts and catalytic supports. Detailed knowledge of the surface structure at the atomic level is critical for a molecular level understanding of the reaction mechanisms involved, i.e. of the corresponding surface chemistry. This in turn allows for a rational design of a catalyst with an improved catalytic response. Much of the work on surface structure determination has been carried out on SrTiO3 single crystals that are commercially available using electron diffraction. In a parallel effort, the synthesis of SrTiO3 powders consisting of nanoparticles of controlled size, shape and surface structure are being studied. We have developed syntheses that produce monodisperse single-crystal nanocubes and nanotubes of SrTiO3 and are moving to additional metal oxides. The increased surface area of nanoparticulate powders offers more active sites for catalysis and functionalization. Hydrothermal and microwave-assisted synthesis conditions are crucial for the formation of highly crystalline fine particles.
High quality single crystals with well-defined stoichiometries are needed to study the intrinsic properties of these catalysts. For fundamental surface studies the single crystals should be free of structural defects and impurity phases. To grow high quality single crystals, the traveling solvent zone method is used. This method requires precise compositions, high-density starting materials, and proper alignment, temperature, oxidizing/reducing atmosphere, and growth rate. Crystals of magnesium orthovanadate, several centimeters in length, were grown for the first time; these are significantly larger than crystals grown in our laboratory previously by conventional flux techniques. The single crystal growth of Mg3(VO4)2 has been studied along with XPS, Raman, transmission electron microscopy, and adsorption studies of various gases (H2, methyl radicals, and O2). These studies probe the reductive addition (H2 and methyl radicals) and the oxidative elimination (O2) reactions occurring on the surface.
Collaborative Centers
Institute for Catalysis in Energy Processes
(http://www.northwestern.edu/catalysis/icep.html)
Institute for Atom-Efficient Chemical Transformations
http://www.anl.gov/catalysis-science/)
Solid oxide fuel cells (SOFC) convert chemical energy to electrical energy, unlike an engine, which uses thermal energy (heat produced from combustion). This chemical process allows for high theoretical efficiencies and fuel flexibility. Hydrogen is an attractive fuel owing to its sole by-product being water. Commercial SOFC units are currently used to power individual buildings and can be designed to power a block of houses. Ni-YSZ cermets are commonly used for SOFC anodes because of their excellent electrochemical performance in hydrogen fuel. However, nickel is susceptible to sulfur poisoning and carbon coking, which are detrimental to anode performance. In order to avoid the challenges associated with Ni metal, several groups have studied conducting oxide materials for application as SOFC anodes. The most successful anodes, in terms of electrochemical performance, have been mixed oxygen-ion and electronically conducting oxides.
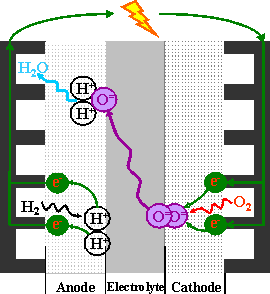
We have tested lanthanum strontium iron chromium oxide (LSFeCr) as an anode and have evaluated it with two separate electrolytes, gallium doped ceria (GDC, Gd0.1Ce0.9O2-δ) and La0.9Sr0.1Ga0.8Mg0.2O3-δ (LSGM) with a La0.6Sr0.4Co0.2Fe0.8O3-δ-GDC cathode. Comparing LSFeCr to La0.8Sr0.2CrO3-GDC anodes, we show that the addition of iron to the structure greatly increased the power density. This arising from the much higher ionic conductivity of oxygen between the FeO3 octahedra and FeO2.5 pyramids, as well as the transport of the oxygen ions bridging FeO3 and CrO3 octahedra. Also attributed to better ionic conductivity is a decreased polarization.
Present and ongoing work in this area is addressing electrode poisoning, namely from an increase in sulfur owing to impurities in the fuel gas (e.g. H2S). This is important owing to a possible wide variety of qualities of fuel and allowing fuel flexibility in real-world applications. The performance of the LSFeCr anode as a function of stoichiometry is also of interest to optimize the anode for chemical stability and electrical performance.