Maria C. Curet-Arana, Randall Q. Snurr and Linda J. Broadbelt
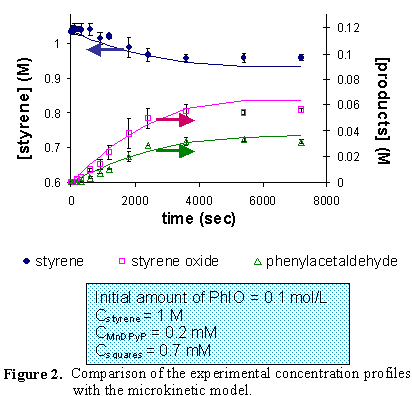
Experiments have performed with free manganese porphyrin systems and with the porphyrins inside the molecular squares as catalysts for the epoxidation of styrene using iodosylbenzene as the oxidant. Two different free porphyrin systems were studied (Mn tetraphenyl porphyrin and Mn dipyridyl porphyrin) in order to compare these systems with the performance of the molecular squares. Results indicate that the catalyst is more stable when it is encapsulated in the molecular squares. Complete conversion of the limiting reactant was obtained, and the turnover number increased 100 times compared to the free porphyrin systems.
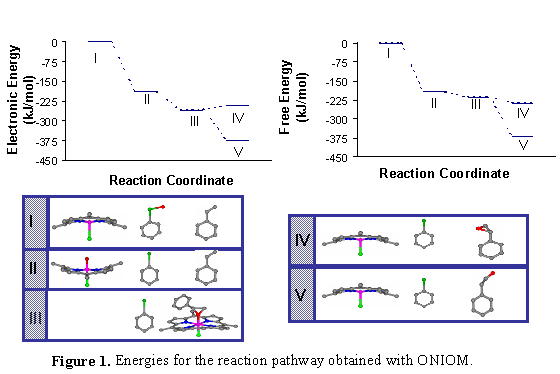
Quantum mechanical calculations have been carried out to obtain information about reaction energetics as a first step in quantifying rate constants for the reaction mechanism. Density functional theory (DFT) and quantum/classical ONIOM calculations have been performed in order to obtain optimized geometries for MnTPP and the intermediates along the proposed reaction path. For the DFT calculations and for the high-level part of ONIOM, the PW91 exchange-correlation functional was used, along with the LANL2DZ basis set, which uses effective core potentials. UFF was used for the low-level part of the ONIOM calculations. Different spin states of the porphyrin and the intermediates were analyzed with both levels of theory. The results from the quantum chemical calculations were incorporated into a microkinetic model that was able to capture the concentration dependence observed experimentally for both the reactant and the catalyst.
Department of Chemical Engineering, Northwestern University