 |
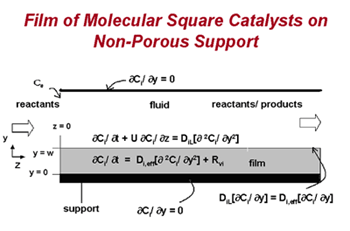 |
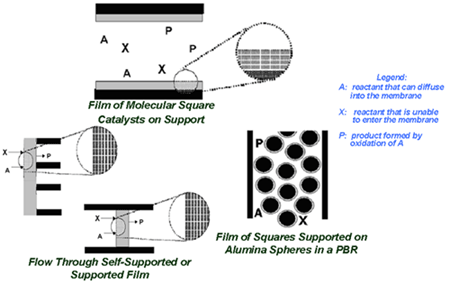
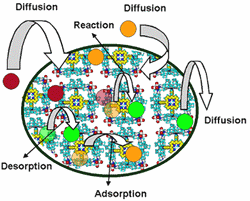
To date, different methods for solving continuum models of styrene epoxidation using molecular square assemblies as catalysts have been developed and explored. Three different reactor configurations have been conceived, and one has been studied in detail. Based on the results of this study, the proposed research will initially focus on applying the most efficient of the solution methods examined to the other reactor configurations to determine the optimal one. However, the analysis that has been performed to date uses an analytical expression for the rate equation derived from experimental data for a homogeneous system. To incorporate more detail about the chemistry, including information about reactive intermediates, two different multiscale strategies will be developed. The first is serial and uses kinetic and transport parameters obtained from quantum chemical calculations and molecular simulations. This is the most straightforward way to incorporate a microkinetic description of the kinetics while retaining a continuum-level model. The second multiscale approach developed will be parallel. In this case, multiple layers of continuum models will be used, and molecular-level information will be fed "on-the-fly" to the continuum models using kinetic Monte Carlo as the microscopic simulator. Incorporation of a microscopic simulator will enable the effect of catalyst inhomogenities on the macroscopic behavior to be gauged.